Norsk Farmasihistorisk Selskap
Denne artikkelen er republisert fra Cygnus nr 18, 2011.
Av Marit Aandahl*
* Cand.pham. UiO 1963. Provisor i Bodø 1965–1968. Sykehusfarmasøyt i Bodø, Lillehammer og Sophies Minde Hospital, Oslo 1969-1980. Laboratoriekjemiker Statens Legemiddelkontroll 1980-1986. Registreringsavdeling i Nycomed Imaging (senere GE Healthcare) 1986–2009. Adresse: Kringsjågrenda 31, 0861 Oslo
Nyegaard & Co. (Nyco) ble etablert i 1874 som et agentur for utenlandske leverandører i apotekbransjen. Virksomheten ble etter hvert utvidet til også å omfatte produksjon av farmasøytiske preparater. Firmaet startet tidlig å arbeide med røntgenkontrastmidler i tillegg til andre farmasøytiske preparater og reagenser. Det første røntgenkontrastmidlet var synonympreparatet Urotrast som ble registrert i Norge i 1934. I femtiårene startet arbeidet med utvikling av egne originale røntgenkontrastmidler og Isopaque (metrizoat) ble registrert i Norge i 1961. Det første ikke-ioniske røntgenkontrastmidlet Amipaque (metrizamid) ble registrert i Norge i 1974. Omnipaque (iohexol), det første ikke- ioniske røntgen-kontrastmidlet i bruksferdig form ble registrert i Norge i 1982 og i Storbritannia (UK) i 1983. Omnipaque er fremdeles blant verdens mest solgte ikke-ioniske røntgenkontrastmidler, selv om patentet er utløpt.
Med Isopaque ble arbeidet med registrering og markedsføring av firmaets kontrastmidler utenfor Norge intensivert, i første rekke i Norden og Nord-Europa. Registrering i andre land økte kraftig etter utviklingen av Amipaque. Amipaque ble etter hvert erstattet av Omnipaque som ble søkt registrert over hele verden. Introduksjonen av Amipaque og Omnipaque var en betydelig innovasjon som raskt førte til et stort salg på verdensbasis. Dette ga grunnlag for det videre arbeidet med kontrastmidler. I disse årene utviklet firmaet seg fra å være et lite selskap til å bli en global aktør og verdensledende på kontrastmidler. Den kliniske forskningen som bidro til utviklingen av firmaets kontrastmidler er beskrevet i Norsk Farmaceutisk Tidsskrift (1).
I 1986 endret firmaet navn til Nycomed og virksomheten ble delt i to divisjoner med kontrastmidlene i en egen divisjon, slik at Nycomed Imaging fortsatte arbeidet med disse. Eksportvirksomheten utgjorde mesteparten av omsetningen av kontrastmidlene.
En egen registreringsavdeling (Regulatory Affairs – RA) for kontrastmidlene ble opprettet i Nycomed Imaging i 1987 med cand.pharm. Solveig Andersen som leder i tillegg til 2 medarbeidere. Dette var en periode med stor aktivitet og ekspansjon i firmaet og det gjaldt også registreringsavdelingen. På slutten av 1990 økte antall ansatte til 12 personer og i 1996 til 28 personer.
Registreringsavdelingens oppgaver var å få Omnipaque registrert i nye markeder i Europa og resten av verden, delta i utviklingen av og ha ansvar for registreringen av de nye røntgen-, MR- og ultralyd-kontrastmidlene som firmaet arbeidet med. Avdelingen hadde kontakt med legemiddelmyndighetene i registreringssaker og skulle også følge med i regelverk for registrering av legemidler, spesielt i Europa. Registreringsavdelingen ble planlagt bygget opp ifølge såkalt «stor modell», dvs. at registreringsdokumentasjonen ble utarbeidet i avdelingen på basis av rapporter fra fagavdelingene og i samarbeid med disse. Fagpersoner med kompetanse innen kjemisk, toksikologisk og klinisk dokumentasjon ble derfor ansatt i registreringsavdelingen. De endelige dokumentene ble godkjent av de ansvarlige i fagavdelingene.
Nycomed Imaging hadde i 1986 datterselskap i de nordiske land, England, Belgia og Nederland. Markedsføring i andre land skjedde via salgskontor og agenter. Schering i Tyskland hadde bidratt i utviklingen av Omnipaque og hadde derfor rettigheter til salg av produktet i enkelte land iVest-Europa. Nycomed Imaging startet nå med å opprette datterselskap i flere av disse landene for selv å kunne markedsføre de nye kontrastmidlene som var under utvikling også der. I tillegg ble det opprettet datterselskap og salgskontorer over store deler av verden. Det var viktig for registreringsavdelingen i Oslo å holde kontakt med kollegaer som hadde lokal RA kompetanse. Avdelingen holdt årlige møter i Oslo med representanter fra datterselskapene i Europa hvor det ble orientert om planene for utsendelser fra avdelingen i det kommende år. I tillegg ble det utvekslet informasjon om lokalt regelverk og knyttet nyttige personlige kontakter. I Nord- og Sør-Amerika skjedde registrering og markedsføring av Omnipaque og de senere produkter via et amerikansk firma (Sterling Winthrop) og tilsvarende med et japansk firma (Daiichi) i Japan. Kontrastmiddeldelen av det amerikanske firmaet ble etter hvert kjøpt opp av det norske selskapet. Registreringsavdelingen samarbeidet hele tiden aktivt med registreringsavdelingen i USA.
Registreringsdokumentasjonen ble utarbeidet iflg. gjeldende EU-regelverk for registrering av legemidler (2). Dokumentasjonen ble skrevet på engelsk i henhold til EU dokumentet «Notice to Applicants» (NTA) fra 1987 og senere oppdateringer (3). Dette anga gjeldende krav til innhold og disposisjon for dokumentasjonen og omfattet krav om ekspertrapporter for den farmasøytisk-kjemiske, toksikologiske og kliniske dokumentasjonen. I perioden 1987–1997 ble det arbeidet med 6 nye kontrastmidler som ble godkjent i mange land i Europa og resten av verden. Første kontakt med helse-/legemiddel myndighetene var søknad om å starte klinisk prøvning. Nødvendig dokumentasjon ble utarbeidet av registreringsavdelingen i samarbeid med fagavdelingene. Det samme gjaldt «Investigators Brochure» med informasjon til utprøver.
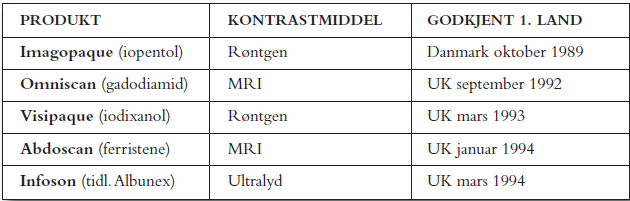
Fem av de nye produktene ble søkt registrert i EU land i Europa via «Multistate» prosedyren (4) som da var mulig i disse landene, slik det fremgår av tabell 1. «Multistate» prosedyren var detaljert beskrevet i NTA 1987 (3). Denne søknadsprosedyren for registrering ble valgt i EU land fordi den anga faste tidsfrister for myndighetene i motsetning til nasjonale søknader som ofte tok lang tid.
I «Multistate» prosedyren ble produktet først godkjent i ett land. Godkjenning og utredningsrapport fra første land ble så brukt som basis for søknad i de andre landene. Selve prosedyren startet når alle land hadde bekreftet mottak av oppdatert dokumentasjon som svar på spørsmål fra 1.land. Prosedyren ble koordinert fra et EU-kontor i Bryssel. CPMP («Committee for Pharmaceutical Medicinal Products») med representanter fra alle EU land anbefalte godkjenning av produktet og et engelsk SPC (Summary of Product Characteristics). Men Markedføringstillatelse (MT), endelig godkjenning av nasjonalt SPC og pakningsvedlegg skjedde i hvert land etter CPMP anbefalingen. Dette innebar at det kunne bli små forskjeller i SPC i de ulike land. Dette var noen av ulempene med denne registreringsprosedyren som i 1995 ble erstattet av gjensidig anerkjennelsesprosedyre («Mutual Recognition Procedure», MRP) (5). I MRP blir det engelske SPC oversatt til nasjonal versjon som godkjennes i hvert av landene som er med i prosedyren.
Danmark ble valgt som første land og «Rapporteur» for Imagopaque, mens UK ble valgt som første land for de fire andre produktene. En kontaktperson hos myndighetene var behjelpelig i løpet av første godkjenning, under oppdatering av dokumentasjonen og i selve «Multistate» prosedyren. Det ble også anledning til å møte representanter for de engelske legemiddelmyndighetene (MCA) for å diskutere søknadene.
Registreringsdokumentasjon utarbeidet på engelsk iflg. NTA var felles for alle land. Søknadsskjema og eventuelle andre dokumenter i tillegg ble utformet iflg. nasjonale krav og på nasjonalt språk om nødvendig. Preparatomtale (SPC), pakningsvedlegg og etikett ble oversatt fra engelsk og om nødvendig justert iflg. nasjonale krav.
Imagopaque ble godkjent i Danmark som første land i oktober 1989. Nycomed Imaging hadde da ikke datterselskap i alle EU land. I Registreringsavdelingen måtte man derfor skaffe seg oversikt over nasjonalt regelverk og ordne de fleste søknadspapirer og oversettelser fra Norge. Den gang var det krav om at deler av dokumentasjonen, bl.a. ekspertrapporter, måtte oversettes til fransk og spansk. Etter hvert ble disse basisdelene akseptert på engelsk i alle EU-land.
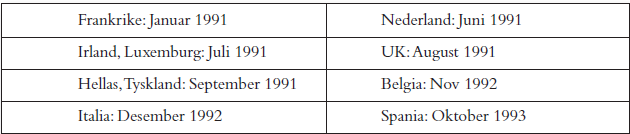
Etter godkjenning i Danmark, måtte utredningsrapporten oversettes til engelsk siden den var utarbeidet på dansk. Etter nødvendig oppdatering av dokumentasjonen og ferdigstillelse av nasjonale søknadspapirer ble søknaden sendt til alle EU-land i mai 1990. Pga. forsinkelse i mottaket i ett land, startet prosedyren i august 1990. CPMP-anbefaling om godkjenning kom 15. mai 1991. Nasjonale godkjenninger kom etter hvert slik det fremgår av tabell 2.
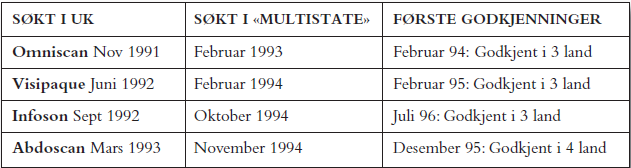
For de neste 4 produktene, Omniscan, Visipaque, Abdoscan og Infoson ble UK valgt som første land og «Rapporteur». Dokumentasjon for flere produkter ble utarbeidet samtidig og nye søknader sendt ut delvis parallelt. Det ble arbeidet mye med å gjøre presentasjon og utforming av dokumentasjonen lett tilgjengelig og oversiktlig i henhold til gjeldende regelverk. Det resulterte i positiv sluttkommentar i den svenske farmasøytiskkjemiske utredningsrapporten for et av produktene: «The content and format of the pharmaceutical dossier is excellent». Mange land krevde flere eksemplarer av deler av filen. En søknad på f.eks. 30 ringpermer som skulle sendes til 10 land, måtte kopieres til 558 permer og ble sendt ut i 100 kartonger i løpet av 5 dager. Tabell 3 viser en oversikt over søknader og godkjenninger (Marketing Authorisation, MA).
Etter nasjonal godkjenning og Markedsføringstillatelse samarbeidet registreringsavdelingen i Oslo med lokale registreringsansvarlige om utarbeidelse av etiketter og pakningsvedlegg iflg. nasjonalt godkjent tekst. Siden dette var flere nye land for firmaet i Europa og etter hvert også utenfor Europa, krevde arbeidet ressurser både i registreringsavdelingen og andre avdelinger som bidro i utarbeidelsen av etikettene som kreves på lokalt språk i de fleste land.
Per 1. januar 1995 kom det nytt regleverk for registreringssøknader i EU. «Multistate» prosedyren ble erstattet av «Mutual Recognition Procedure» (MRP) og «Concertation procedure» ble erstattet av «Centralised procedure» (6). Samtidig ble EMEA (“European Agency for Evaluation of Medicinal Products”) opprettet i London. I den sentrale prosedyre utsteder Kommisjonen i Bryssel en Markedsføringstillatelse som gjelder for alle EU land. Engelsk tekst for SPC, pakningsvedlegg og etikett godkjennes i nasjonal oversettelse for alle land. Det er faste tidsfrister for søker og myndighet i løpet av prosedyren.

Det ble vedtatt at det nye MR kontrastmidlet Teslascan skulle søkes godkjent i EU i den sentrale prosedyren. Irland ble oppnevnt som «Rapporteur» med Nederland som «Co-Rapporteur». Registreringssøknaden ble sendt i juni 1996 og Markedsføringstillatelsen for EU ble utstedt av Kommisjonen i mai 1997, dvs. 11 mnd behandlingstid. Teslascan var produkt nr 40 i EU som brukte den sentrale prosedyren.
Parallelt med «Multistate» og sentral søknad i EU ble de nye kontrastmidlene søkt registrert i EFTA landene. Etter godkjenning i Norge og Irland (produksjonslandene) ble Imagopaque, Omniscan,Visipaque og Teslascan søkt godkjent i aktuelle markeder i Øst-Europa, Midt-Østen, Asia, Kina, Australia og Afrika.
Mange av disse landene krever «Certificate of a Pharmaceutical Product» (CPP) som del av søknaden som dokumentasjon på at produktet er godkjent i opprinnelseslandet.
Etter hvert ble det for noen av produktene utarbeidet dokumentasjon for nye indikasjoner, ny plast emballasje og nye volum som ble søkt godkjent i land der produktene tidligere var godkjent. I tillegg ble det søkt om godkjenning for variasjoner av SPC, variasjoner i de farmasøytisk-kjemiske dokumentasjoner, ny fabrikk i Irland og endring av firmanavn.
Innehaver av Markedsføringstillatelsen («MA-Holder») er i EU-regelverket pålagt detaljert ansvar og oppfølging av produktet og er også kontaktpunkt for legemiddelmyndighetene i det aktuelle EU-land. Det var firmapolicy at «MA-Holder» skulle være firmaet i Oslo der det var mulig og praktisk.
For et produkt godkjent i den sentrale prosedyre blir det én «MA-Holder» felles for alle EU land. Det var derfor viktig at det var firmaet i Oslo som kunne være kontakt til EMEA og legemiddelmyndighetene i disse landene og også angitt som ansvarlig for produktet på etikett og i pakningsvedlegg.
Det var angitt i art. 4 av EU Direktiv 65/65 og art. 2 av Reg. 2309/93 at MA-Holder skal være «established in the Community». Før søknad for Teslascan i den sentrale prosedyre ble EMEA kontaktet om dette, og de ville ikke akseptere firmaet i Norge som MA- Holder i den sentrale prosedyren. Registreringsavdelingen tok derfor saken opp med Kommisjonen i Bryssel med henvisning til EØS («EEA») avtalen. Det ble vist til at Norge pga. den avtalen i denne sammenheng kunne anses som en del av «community» slik at firmaet derfor burde kunne være «MA- Holder». Kommisjonen i Bryssel aksepterte med henvisning til EØS avtalen at «established in the Community» skulle tolkes som «established in the EEA» Dette er nå spesifisert i EU søknadsskjema og denne tolking er tatt med i kap. 1 i gjeldende utgave av NTA. Dette fremgår av fig 1 Kommisjonens brev 10.07.1996 og fig 2 Informasjon fra «Pharmaceutical Committee». Det betydde at firmaet i Oslo kunne være Innehaver av Markedsføringstillatelsen for Teslascan i EU. Teksten fra det gamle regelverket er nå med i art 8 av Direktiv 2001/83/EC og art. 2 av Reg. 726/2004 (erstatter Reg. 2309/93).


Omnipaque ble i denne perioden godkjent og registrert i 51 nye land. Per 1. januar 1998 var produktet registrert i ca 100 land over hele verden. Markedsføringstillatelsen ble overført fra Schering i noen land i Sør- Europa. Variasjoner i SPC og i den famasøytisk-kjemiske dokumentasjonen ble søkt i land der produktet tidligere var godkjent i Vest- og Øst-Europa, Canada, Midt-Østen, Asia, Kina, Australia og Afrika. Ny plast emballasje og nye volum i glass og plast ble også søkt og godkjent i de samme markeder. I tillegg ble det søkt om godkjenning for ny fabrikk i Irland og endring av firmanavn.
Registreringsavdelingen i Oslo arbeidet som tilsvarende avdelinger i internasjonale farmasøytiske firmaer både i nasjonale og EU prosedyrer. Søknadsprosessen i EU via EU prosedyrer ble enkelt håndtert for firmaet basert i Norge. I den sentrale prosedyren ble det samarbeidet med EMEA som hadde større ressurser, og det ble anledning til å diskutere søknader med dem.
Etter hvert ble det enklere å følge med i EU- regelverk fra Kommisjonen, EMEA og fra andre land via nasjonale hjemmesider på nettet. Det var også nyttig for avdelingen at ansatte fikk delta i internasjonale registreringskurs i Europa om registrering i EU og andre deler av verden. Godt samarbeid mellom lokale og sentrale registreringsavdelinger var viktig for å oppnå godkjenning av søknadene.